Database
implementation
The schematic structure of the ECRbase
data analysis is presented in Figure S1. The database first processes whole genome pairwise
alignments of multiple vertebrate genomes available from the ECR Browser to
identify Evolutionary Conserved Regions (ECRs).
Currently there are over 26M ECRs available
in the ECRbase that correspond to regions extracted
from pairwise comparisons of all the available
species. Next, these ECRs are used to
determine synteny blocks that interconnect these genomes.
Since the identified synteny blocks are based
on nucleotide alignments, not on protein similarity, are thus capable of more
accurately demarcate synteny breakpoints in long intergenic regions. This strategy potentially provides user with
more accurate synteny maps with longer syntenic
stretches for closely related vertebrates (such as human and mouse, for
example) than those restricted to gene comparisons.
In parallel to the ECR identification we've
implemented the extraction of vertebrate promoters using RefSeq,
knownGene, and other species RefSeq
gene annotations available from the UCSC Genome browser database
(Hinrichs et al. 2006,Pruitt et al. 2005). At the
final step, DNA sequences for the identified ECRs and promoters undergo
annotation of TFBS. Subsequently, all
the processed data is collected, binned according to the corresponding genome,
and distributed through the central ECRbase interface
available at http://ecrbase.dcode.org.
Large ECRs and TFBS files are compressed (using the gzip
utility) to facilitate data download. Despite the compression, some of the files continue to be relatively
large and, therefore, some users may find it helpful to use automated file
download utilities for fetching data from the ECRbase.
Below we summarize the details of methods
employed for data extraction and generation.
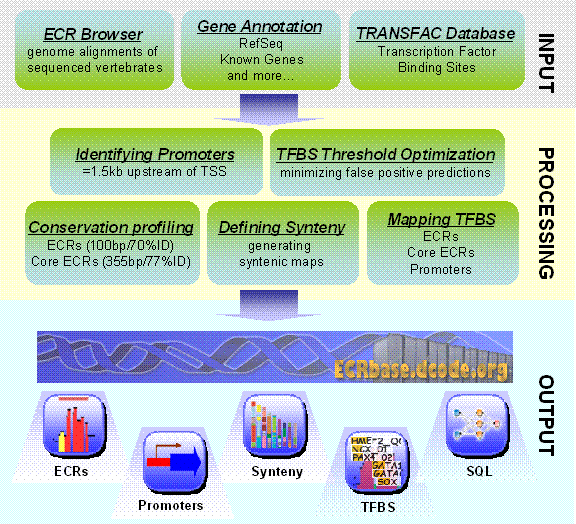
Fig.
1. Schematic pipeline for the ECRbase data analysis.
Evolutionary Conserved Regions.
ECRs are computed as regions
greater than 100bps in length and greater than 70% nucleotide sequence identity
(Table S1). For a region to be
classified as an ECR, it is required to be present in both species. There are cases when a conserved region in
one species has accumulated significant insertions in the second species and therefore,
its second species conservation falls below the required threshold. Elements that exhibit this conservation
pattern are excluded from the database.
Stricter thresholds, of a minimum length of 350bps and conservation
level of 77% ID are used for identifying conserved elements termed coreECRs –
regions that are implied to have a higher probability of being functional than
regular ECRs (Ovcharenko et al.
2004,Prabhakar et al. 2006). ECRbase reports genome positional information of ECRs (and
coreECRs), their length and percent identity as well as the corresponding
parameters for their orthologs in other genomes.
Table S1. Number of ECRs in inter-species alignments for the
human (hg18), dog (canFam2), mouse (mm8), rat (rn4), chicken (galGal3 & gg2), frog (xt4),
and Fugu (fu4) genomes (in thousands)
|
|
Dog |
Mouse |
Rat |
Chicken |
Frog |
Fugu |
|
Human |
2,521 |
1,289 |
1,189 |
200 |
120 |
73 |
|
Dog |
|
1,042 |
972 |
178 |
115 |
71 |
|
Mouse |
|
|
2,311 |
169 |
109 |
74 |
|
Rat |
|
|
|
162 |
107 |
70 |
|
Chicken |
|
|
|
|
117 |
67 |
|
Frog |
|
|
|
|
|
73 |
Synteny.
Synteny between vertebrate genomes is determined as
previously described (Ovcharenko et al. 2005). Briefly, we use sets of 3 consecutive ECRs
(two neighboring ECRs are selected as ‘consecutive’ if they are separated by
<100kb in both genomes) to define anchors of inter-genome synteny. These synteny anchors are used to construct
larger synteny blocks by clustering ECR triplets from matching chromosomes
using the same maximum 100kb separation threshold (Table S2). Since a great number of genomes are available
in draft sequence format (in a multi-scaffold configuration), several
artificial synteny breakpoints originate simply from the scaffold edges
prematurely disrupting the syntenic structure. Short scaffolds can also potentially prevent
the identification of the 3-ECR synteny anchors thus also leading to the
elimination of some synteny relationships and/or generation of incomplete syntenic blocks. For
that reason, synteny assignments originating from unfinished genomes should be
treated with caution.
Promoters.
ECRbase utilizes RefSeq, knownGene, and ‘other
species RefSeq’ gene annotation available at the UCSC
Genome browser database (Hinrichs
et al. 2006,Pruitt et al. 2005) to localize the genomic position and the strand
of gene transcripts in vertebrate genomes.
Overlapping transcripts are combined into unique genes and the outermost
5’ end is used as a landmark of transcriptional start site (TSS). Alternative promoters that are exonic, intronic or in untranslated regions can not be determined by this analysis
and are therefore not represented in the database. Next, the data extraction utility selects the
1.5kb region upstream of the gene TSS, annotates it as the promoter element and
automatically fetches the corresponding DNA sequence (repetitive elements are
indicated by lower-case letters consistent with data representation in the UCSC
Genome browser). Promoter elements are
limited to intergenic spaces and are dependent on the location of neighboring
genes. In cases where the intergenic
region is significantly shorter than 1.5kb, the identified promoters span the
entire intergenic space between the two transcripts and are therefore less than
1.5kb. ECRbase
reports positional and directional information of promoters as well as it
provides the name of the gene the promoter is associated with. Bi-directional promoters (promoters shared by
two genes transcribed in a head-to-head manner) are reported twice – once for
each transcript.
Table S2. Longest syntenic block size
from inter-species comparison of the human (hg18), mouse (mm8), chicken (gg2),
frog (xt4), and Fugu (fu4) genomes
(in thousand basepairs, kb)
|
Second |
Human |
Mouse |
Chicken |
Frog |
Fugu |
|
Human |
- |
56,101 |
48,475 |
9,233 |
9,656 |
|
Mouse |
54,507 |
- |
38,134 |
7,888 |
8,783 |
|
Chicken |
19,105 |
16,106 |
- |
7,952 |
4,704 |
|
Frog |
5,479 |
5,333 |
6,529 |
- |
3,529 |
|
Fugu |
1,990 |
1,723 |
1,539 |
1,397 |
- |
Transcription factor binding sites.
We utilize TRANSFAC Professional database of PWM
corresponding to vertebrate transcription factors (version 9.4) (Matys
et al. 2006)
to map candidate TFBS in genomic sequences.
TFBS are mapped as previously reported, using the tfSearch
(Ovcharenko et al. 2005) utility that employs a suffix
tree technique to rapidly identify motifs in DNA sequences. In an effort to limit the number of false
positive TFBS predictions we avoid using default PWM sequence similarity
parameters, but instead perform an independent optimization of thresholds for
different TFBS that warrants 5 or less TFBS predictions per 10kb of random
sequence. Each ECR and promoter element
undergoes a TFBS mapping, and positional and directional information of each
TFBS inside these elements. TFBS data is
next collected and distributed through the corresponding portal of the ECRbase.
References.
Ovcharenko, I., Nobrega, M.A., Loots, G.G. and Stubbs, L. (2004) ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res, 32, W280-286.
Hinrichs, A.S., Karolchik, D., Baertsch, R., Barber, G.P., Bejerano, G., Clawson, H., Diekhans, M., Furey, T.S., Harte, R.A., Hsu, F. et al. (2006) The UCSC Genome Browser Database: update 2006. Nucleic Acids Res, 34, D590-598.
Pruitt, K.D., Tatusova, T. and Maglott, D.R. (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res, 33, D501-504.
Ovcharenko, I., Stubbs, L. and Loots, G.G. (2004) Interpreting mammalian evolution using Fugu genome comparisons. Genomics, 84, 890-895.
Prabhakar, S., Poulin, F., Shoukry, M., Afzal, V., Rubin, E.M., Couronne, O. and Pennacchio, L.A. (2006) Close sequence comparisons are sufficient to identify human cis-regulatory elements. Genome Res, 16, 855-863.
Ovcharenko, I., Loots, G.G., Nobrega, M.A., Hardison, R.C., Miller, W. and Stubbs, L. (2005) Evolution and functional classification of vertebrate gene deserts. Genome Res, 15, 137-145.
Matys, V., Kel-Margoulis, O.V., Fricke, E., Liebich, I., Land, S., Barre-Dirrie, A., Reuter, I., Chekmenev, D., Krull, M., Hornischer, K. et al. (2006) TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res, 34, D108-110.
Ovcharenko, I., Loots, G.G., Giardine, B.M., Hou, M., Ma, J., Hardison, R.C., Stubbs, L. and Miller, W. (2005) Mulan: multiple-sequence local alignment and visualization for studying function and evolution. Genome Res, 15, 184-194.